
Ethics and study design
Table of Contents
- Ethics and study design
- Participants
- Sample collection
- Proteomic data generation using the Olink Explore 3072 assay
- Genetic data generation
- Cohort sampling for the Discovery/Training and Replication/Testing datasets
- Statistical analysis of the individual protein analytes
- Comparison of Olink and ELISA for differentially abundant proteins
- Comparison of Olink and SomaScan data for plasma proteins
- Comparison of protein in CSF and plasma
- Pathway analysis
- Regional genetic association analysis and Mendelian randomization
- Supervised machine learning
- SHAP values and ALS risk score
- Inclusion and ethics statement
- Reporting summary
This study was a cross-sectional analysis of plasma proteomic data to identify a biomarker panel for ALS. Samples were obtained from participants from the United States and Italy. Recruitment sites were located at the University of Turin in Turin, Italy; the NIH in Bethesda and Baltimore, Maryland; and Johns Hopkins University in Baltimore, Maryland.
Written consent was obtained from all individuals enrolled in this study. The institutional review boards of the National Institute on Aging (NIA) (protocol numbers 03-AG-0325 and 03-AG-N329); the National Institute of Neurological Disorders and Stroke (01-N-0206, 13-N-0188 and 17-N-0131); the National Institute of Allergy and Infectious Diseases (NIAID) (09-I-0032); Johns Hopkins University (00173663); and the University of Turin (004462) approved the study.
Participants
From September 2008 through February 2023, 281 patients with ALS and 258 healthy individuals were enrolled in the study at the University of Turin and the NIH. The Italian samples consisted of neurologically healthy individuals (n = 196) and patients diagnosed with ALS (n = 236) living in Northern Italy and recruited in a population-based study known as the Piedmont and Valle d’Aosta Registry for ALS (PARALS; established 1 January 1995)9. The registry’s near-complete case ascertainment of ALS among its catchment population of almost 4.5 million inhabitants ensures the applicability of our findings9. The US plasma samples comprised patients diagnosed with ALS (n = 45) evaluated at the NIH Clinical Center in Bethesda, Maryland, as part of a natural history study (NCT03225144)47 and control samples collected as part of the NIH Baltimore Longitudinal Study of Aging (BLSA) (n = 53, Nct00233272)48 and at the Johns Hopkins Hospital (n = 9). The control samples for the CSF analyses (n = 89) were collected under an NIAID natural history protocol (Nct00794352).
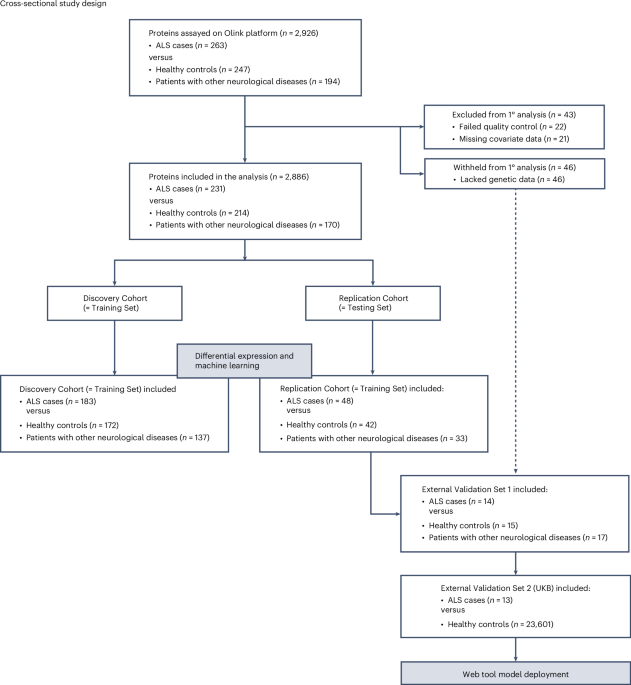
Figure 1 shows the study workflow. The patients with ALS were diagnosed according to the revised El Escorial criteria10 by a neurologist specializing in ALS. Patients with familial and sporadic ALS and self-declared diverse ancestry (n = 3 Hispanic samples) were included in the study, ensuring a comprehensive representation of the ALS population. The control individuals were selected based on their lack of a diagnosis of ALS, neurological disease and cognitive decline in their clinical history. The control cohorts were matched to the case cohorts for race and ethnicity but not for sex or age, although the age distribution was similar (Extended Data Table 1).
We also assembled a cohort of individuals with other neurological conditions (n = 194, listed in Extended Data Table 1) as an additional comparison cohort. Plasma samples were collected from these participants at the NIH Clinical Center (n = 172) and Johns Hopkins University (n = 22). The other neurological disease cohort included patients diagnosed with the following conditions: corticobasal syndrome (n = 8 patients) was diagnosed according to the Armstrong criteria49; patients with Lewy body dementia (n = 8) were diagnosed with clinically probable disease according to consensus criteria50,51; the multiple system atrophy (n = 5) cases were diagnosed according to the Gilman criteria52; and Parkinson’s disease (n = 153) and progressive supranuclear palsy (n = 19) were diagnosed based on the 2015 and 2017 Movement Disorders Society criteria, respectively53,54. One patient was labeled as having dementia, not otherwise specified, after clinical evaluation.
Sample collection
The plasma samples were collected via phlebotomy of the upper limb. The patients were not fasting. The Italian samples (n = 427) were mainly collected using heparin tubes, and the remaining samples (n = 306) were collected using EDTA tubes. Blood cells were removed by centrifugation within 2 hours of collection, and the supernatant, consisting of the plasma, was carefully removed without disturbing the cell pellet. All plasma samples were aliquoted and stored at −80 °C. The number of freeze–thaw cycles between aliquoting and running the proteomic assays was minimized, typically one but no more than three.
The CSF samples (n = 14) were collected via lumbar puncture with the patient sitting upright. A 22-gauge Sprotte spinal needle was used and inserted at the L3/L4 level. The CSF was collected in polypropylene tubes, immediately placed on ice and sent to the laboratory for processing within 30 minutes. All samples were gathered at a single location, the NIH Clinical Center in Bethesda, Maryland (NCT03109288).
Proteomic data generation using the Olink Explore 3072 assay
For each study participant, we performed proteomic profiling on the plasma samples using the Olink Explore 3072 assay (Thermo Fisher Scientific) at the Laboratory of Clinical Investigation at the NIA, according to the manufacturer’s protocol. The Olink Explore 3072 assay quantifies 2,926 proteins (https://olink.com/products/olink-explore-3072-384) with high accuracy and specificity11. In brief, the platform is based on oligonucleotide-labeled antibody pairs that bind to their target protein in solution11. When both antibodies are close, their oligonucleotide tails hybridize and undergo extension by a DNA polymerase. This molecular process generates a unique, double-stranded DNA barcode, which is subsequently amplified and quantified using next-generation sequencing (Illumina NovaSeq).
We implemented quality control, normalization and data calibration at the probe, sample and plate levels11. Inter-plate sample controls (n = 2 per 96-well plate) were used to normalize for inter-plate variation. Proteins that did not meet the default standard Olink quality control criteria were excluded from the analysis. These steps were conducted using Olink software (version 1.0). Normalized Protein eXpression (NPX) values, representing the log2-transformed ratio of sample assay counts to extension control counts, were used as the final assay readout; a higher value corresponded to a higher protein expression or abundance levels11. Replicate samples (n = 3 per plate), referred to as bridging samples, were used to calculate both intra-plate and inter-plate coefficients of variation. These bridging samples are independent of the inter-plate sample controls. The coefficients of variation (%) were determined using the following formula: (s.d./mean of the replicate measurements) × 100. Samples with median NPX values (calculated from all proteins in each sample) that deviated by more than 3 s.d. from the mean of the median NPX values of all samples, as well as samples exceeding 3 s.d. from the mean of the interquartile range, were flagged as outliers and excluded from the analysis.
Olink data from the UK Biobank were obtained through the UK Biobank Research Analysis Platform (https://www.dnanexus.com/partnerships/ukbiobank). ALS cases were defined as individuals diagnosed with ICD-10 code G12.2 and confirmed at death55 and had their blood collected up to 1 year before symptom onset. Control samples were defined as individuals without a recorded diagnosis of myopathy (ICD-10 codes G70–G73) or neuropathy (ICD-10 codes G60–G64).
Genetic data generation
DNA obtained from the same participants also underwent whole-genome sequencing on a HiSeq X10 sequencer (Illumina; n = 476, 150-bp paired-end reads, 35× coverage)56 or genotyping on Infinium GDA-8+ NeuroBooster BeadChips (version 1.0, Illumina; n = 245), according to the manufacturer’s instructions. Standard sample-level and variant-level quality control procedures were applied to the genetic datasets, and variants were extracted for the principal component analysis, performed using ‘flashPCA’ (version 2.0). Principal components 1–10 were reduced to two dimensions using UMAP version 0.2.10. These two UMAP values were used to correct for population stratification in the statistical analysis of the individual protein analytes57.
Although methods such as ADMIXTURE can provide percentage estimates of ancestry, the UMAP approach offers a direct and efficient way to control for confounding in our association analyses. Notably, reanalysis using all ten principal components yielded nearly identical results, confirming that the two-component UMAP adjustment sufficiently controlled for stratification in our study cohorts.
Cohort sampling for the Discovery/Training and Replication/Testing datasets
Samples that passed Olink’s quality control were divided into two cohorts at an 80:20 ratio, labeled as the Discovery/Training and Replication/Testing datasets. The terms ‘Discovery’ and ‘Replication’ refer to differential protein abundance analysis, whereas ‘Training’ and ‘Testing’ pertain to machine learning analysis. To accurately represent cohort characteristics in the datasets, samples were matched by diagnostic groups (ALS, healthy controls and neurological controls) and the type of tube used for collection, as implemented in the ‘caret’ software package (version 6.0-94). We also balanced the selection of the Italian and United States samples across the Discovery and Replication cohorts. The Training Set included 492 individuals (n = 183 ALS cases, n = 172 healthy controls and n = 137 individuals with other neurological conditions). The Testing Set included 123 individuals (n = 48 ALS cases, n = 42 healthy controls and n = 33 individuals with other neurological diseases) (see Fig. 1 for the flowchart).
Statistical analysis of the individual protein analytes
We performed a cross-sectional association analysis between plasma protein abundances and the diagnosis of ALS. The following generalized linear model was used to evaluate the relationship between an individual protein’s abundance and ALS status: NPXi = b1 × asi + b2 × agei + b3 × sexi + b4 × tubetypei + b5 × Map1i + b6 × Umap2iwhere NPXi represents the NPX value for individual i; Wheni is a binary indicator for ALS diagnosis (1 for ALS, 0 for control); agei is the age at sample collection; sexi is the reported sex of the individual; tubetypei indicates the plasma collection tube type (heparin or EDTA); and UMAP1i and UMAP2i are the first two components of the UMAP projection of genetic data. The absence of a constant term means that there is no intercept (b0) in the model. This model was implemented using the ‘limma’ package (version 3.58.1) in R. In our analysis, all covariates were included in the model simultaneously. In alignment with our primary analysis, the regression model for the patients carrying the C9orf72 expansion was adjusted for sex, age at sample collection, the type of plasma collection tube and the first two dimensions of UMAP based on genetic data.
Multiple comparisons were controlled using the FDR procedure defined by Benjamini and Hochberg, and two-sided FDR-adjusted P values are reported in Table 1Fig. 2 and Extended Data Table 2. The significance threshold was set at an FDR P value of 0.05. Our Discovery Cohort, comprising 183 cases and 309 controls, had 80% power to detect a differentially abundant protein with an effect size (Cohen’s f statistic) of 0.242, assuming a significance threshold P value of 1.73 × 10−5. All analyses were conducted using R (version 4.3.2).
We used z-scores to compare the effect sizes of differentially abundant proteins in the Discovery and Replication cohorts. We used z-scores in this comparison because they provide an accurate statistical representation of the difference in effect size between cases and controls by taking into account the s.e. of the effect size58. Without standardizing with z-scores, the slope of the relationship between protein abundance and disease status could be artificially inflated or deflated, resulting in misleading correlation statistics.
Comparison of Olink and ELISA for differentially abundant proteins
Quantification was performed on plasma samples (n = 16 ALS cases and n = 16 healthy controls) using commercially available colorimetric ELISA or fluorescence-based ProQuantum high-sensitivity immunoassay kits according to the manufacturer’s protocol. Immunoassay kits were available for 30 of the 33 differentially abundant proteins (Extended Data Fig. 1). The optimal dilution factor for measuring each protein was determined empirically. Each plasma sample and protein standards were assayed in duplicates. A standard curve for each protein was generated by plotting the absorbance readings at 450 nm wavelength or FAM fluorescence emission at 517 nm against the concentrations of the protein standards. A four-parameter logistic curve was fitted using GraphPad Prism software (version 10.4.1). The protein concentrations for the test samples were extrapolated from the standard curve, and the final concentrations were corrected for the dilution factor.
The Bland–Altman plot was created by calculating the differences in measurements between the ELISA or ProQuantum and Olink assays (log2-transformed concentration from the ELISA or ProQuantum assay minus Olink NPX) and comparing this to the mean measurement from both assays for each paired sample. This standard data visualization technique is recommended for comparing methods59. The 95% limits of agreement were calculated using the mean difference (bias) ± 1.96 s.d. of the differences. The relationship between the protein measurements from both assays was evaluated using the Pearson correlation coefficient. For the subset of samples (n = 16 ALS and n = 16 healthy controls), differential protein abundance analysis was performed using a generalized linear regression model with the following formula: ProteinAbundancei = b1 × ifi + b2 × agei + b3 × sexi + b4 × tubetypei. Here, ProteinAbundancei denotes the protein NPX value measured via Olink or the log2-transformed concentration from the ELISA or ProQuantum assay for individual i; Wheni is a binary variable for ALS diagnosis (1 for ALS, 0 for control); agei refers to the age at which the sample was collected; sexi indicates the self-reported sex of the individual; and tubetypei specifies the type of plasma collection tube (heparin or EDTA). The intercept was set to 0. All analyses and visualizations were conducted using GraphPad Prism software (version 10.4.1).
Comparison of Olink and SomaScan data for plasma proteins
Proteomic measurements were previously generated for a subset of the BLSA control samples (n = 9) using SomaScan 7K, a single-stranded DNA aptamer-based proteomic platform60. To assess the correlation, the log2-transformed NPX values for the Olink 3072 Explore platform and the scaled measurements for the SomaScan 7K platform were used to calculate Spearman’s rho correlation for the 2,868 unique UniProt proteins assayed on both platforms (Extended Data Fig. 2A)11. The data were categorized using the k-means clusters identified by Katz et al.11 (Extended Data Fig. 2B).
Comparison of protein in CSF and plasma
CSF proteomic measurements were generated from ALS cases (n = 14) and healthy control individuals (n = 89) using the SomaScan 7K platform. Most of the ALS cases (n = 13) and none of the healthy control individuals used to generate the SomaScan data were not included in the Discovery Cohort or the Replication Cohort used to create the plasma proteomic data. Individual proteins were assessed using generalized linear modeling, adjusted for age at collection and sex.
Pathway analysis
The proteins that were differentially abundant in ALS were analyzed for pathway enrichment using ‘clusterProfiler’ (version 4.10.1) using the following databases: (1) GO61(2) KEGG62 and (3) Reactome63. Pathways with a single gene were discarded, and significance was defined as an FDR-adjusted P value of less than 0.05.
Regional genetic association analysis and Mendelian randomization
Regional association plots of the genes encoding the differentially abundant proteins identified in ALS cases were made with ‘locusZoomR’ (version 0.3.5) using data downloaded from a large ALS GWAS17. Single-nucleotide polymorphisms (SNPs) showing a P value less than or equal to 5.0 × 10−8 were regarded as evidence of genetic variants in the locus that may influence the risk of developing ALS.
We performed Mendelian randomization analyses using ‘TwoSampleMR’ (version 0.6.1)64. Exposures were the pQTL mapping of the plasma proteins identified by the machine learning model. These plasma pQTL profiles were obtained from GWAS data based on 54,219 UK Biobank participants, available from the Synapse web portal18. Only genetic variants with P < 1.0 × 10−5 from the pQTL summary statistics were included in the analysis. As the outcome, we used an ALS GWAS, comprising 29,612 cases and 122,656 control individuals17. To assess causality, we applied the inverse‐variance weighted method and two Mendelian randomization sensitivity tests (MR Egger and weighted median). Traits were considered consistent with a causal effect when P values were less than or equal to 0.05.
Supervised machine learning
We performed supervised machine learning on the proteomic data to identify a molecular signature of ALS. To do this, we randomly selected 80% of the samples as the Training Set (n = 183 ALS cases versus n = 172 healthy controls plus n = 137 other neurological diseases). The remaining samples were designated as the Testing Set (n = 48 ALS cases versus n = 42 healthy controls plus n = 33 other neurological diseases; see Fig. 1 for flowchart). The samples in the Training Set and the Testing Set were the same as those in the Discovery Cohort and the Replication Cohort used for the differential protein abundance analysis, respectively.
We used ‘caret’ (short for ‘classification and regression training’, version 6.0-94), an open-source machine learning package that streamlines predictive model creation. The 33 differentially abundant proteins, along with clinical information, such as age at sample collection, sex and the type of blood tube used to collect the sample (total number of features = 36), were entered into the model as the starting features.
The decision to limit the feature set to 33 proteins was made to enhance the model’s stability and avoid overfitting, often seen in high-dimensional data analysis65. Additionally, using a minimal feature set makes the model more relevant in clinical environments, where cost and assay complexity play important roles. For instance, even though UMAP components are useful for addressing population stratification in statistical analyses, they were left out of the machine learning model. Their inclusion would require all patients to undergo genotyping, substantially increasing implementation costs and computational complexity. Consequently, we concentrated on proteins that can be directly measured in plasma, ensuring that the model remains both practical and affordable.
Ten supervised machine learning algorithms were evaluated (listed in Extended Data Table 5). Fivefold cross-validation was conducted and repeated ten times, and data were preprocessed with centering and scaling to standardize features. Adaptive resampling with a minimum of ten resamples, and a significance threshold of a = 0.05 using generalized least squares was employed to refine the tuning parameters for each model. Models were trained on 36 features, using the sample diagnosis as the binary target variable. The best model (random forest) was selected based on overall performance (AUC and balanced accuracy) when classifying unseen samples in the Testing Set, External Validation Set 1 and External Validation Set 2.
The random forest model was further tuned on the Training Set, and key predictive features were prioritized using the genetic algorithm feature selection method; fivefold cross-validation was conducted and repeated ten times, and data were preprocessed with centering and scaling to standardize features. The parameters used for finetuning and feature selection using the genetic algorithm method were as follows: nbits = 36, popSize = 500, maxiter = 25, pmutation = 0.1, pcrossover = 0.8 and elitism = 3. The hyperparameter for the optimized random forest model, ‘mtry’ (the number of variables considered at each split), was set to 11. For model tuning in the Discovery Cohort, the standard random forest majority-vote threshold of 0.5 was used and then directly applied to the Replication Cohort. The final model was selected based on the performance in the Testing Set, External Validation Set 1 and External Validation Set 2 cohorts.
We conducted a repeated classification analysis, excluding NEFL, to assess the impact of this protein on the model’s performance. The remaining features (that is, 16 proteins, demographic factors (sex and age at sample collection) and plasma collection tube type) were incorporated into this model.
SHAP values and ALS risk score
SHAP values were calculated using the Shapley function from the ‘iml’ package (version 0.11.3) in R. These values reflect the contribution of each feature to the final random forest model. To determine the ALS risk score, we summed the SHAP values for all features (n = 20, including 17 proteins, sex, age at sample collection and type of tube used for collection) for each sample. The summed SHAP values were then scaled to a range from 0 to 100 based on the minimum and maximum values from the Training Set.
Subsequently, we conducted a generalized linear regression analysis, adjusting for age at sample collection, sex and plasma tube collection type, to assess the correlation between the time to symptom onset and (1) the ALS risk score and (2) individual protein abundances.
Inclusion and ethics statement
All collaborators of this study who have fulfilled the criteria for authorship required by Nature Portfolio journals have been included as authors, as their participation was essential for the design and implementation of the study. Roles and responsibilities were agreed among collaborators ahead of the research. This work includes locally relevant findings, which have been determined in collaboration with local partners. This research was not severely restricted or prohibited in the researchers’ setting and did not result in stigmatization, incrimination, discrimination or personal risk to participants. Local and regional research relevant to our study was considered in citations.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.